Drug Substance Solid State Characterization
Introduction
A thorough knowledge of an active pharmaceutical ingredient’s (API) solid state behavior is essential for reliable drug product manufacturing, patent protection, and formulation viability. This article discusses various aspects of the API in the solid state which can impact pharmaceutical product development.
Solubility and Permeability
Solubility and permeability dictate bioavailability and thus dosage form efficacy. The Biopharmaceutical Classification System (BCS) classifies compounds based on potency, solubility and membrane permeability, providing a guide for formulation development1. A first step in preformulation is to determine the API’s pH solubility profile. For ionizable compounds, these results establish the relationship between charge state and pH; the ionization constant (pKa) allows prediction of a compound’s absorption and distribution in vivo. Another useful property is the partition coefficient (log P), which describes drug partitioning between aqueous and organic phase; this is commonly measured with chromatographic methods. Knowledge of pKa value(s) and log P are critical to understanding the ability of an ionized molecule to permeate biological membranes2.
The majority of drugs in development and those recently launched are poorly water soluble; this presents a particular challenge in designing an optimal formulation3. Ways to improve solubility include chemical modification of APIs (e.g., prodrugs), use of co-solvents or other excipients (surfactants, phospholipids, cyclodextrins and other polymers), and manipulation of particle size/ morphology. For the latter, there has been a growing trend to apply particle size reduction and to control particle morphology. Reduction of particle size, most commonly through (nano)-milling, increases specific surface area, leading to enhanced dissolution and improved homogeneity of the bulk material. Finally, selection of the API solid form can also significantly influence solubility and thus formulation performance.
Salts
Acid or base drugs can often be converted to salts, resulting in solubility improvement4. Salt screening is generally done in discovery to identify the best API form(s) for development; the drug is exposed to counterions and crystallization solvents under various conditions. Products are typically analyzed by powder X-ray diffraction (XRD) and/or Raman spectroscopy, to confirm “hits” for further study and scale-up. And while salts are a potential avenue to explore, the toxicology and salt stability have to be considered and evaluated prior to moving forward. For formulation, the toxicology considerations depend on the type of salt and the planned dose. As to the stability, the salt integrity in the dosage form must be maintained to avoid reconversion to the less soluble acid or base form.
Polymorphs
Many APIs can exist in multiple crystal forms, i.e., chemically identical but physically distinct as solids. There is also crystal habit, wherein a polymorph exhibits an external shape, conferring distinct and possibly desirable properties5. Early polymorph screening identifies and selects the best forms(s) for further evaluation. The existence of drug polymorphs has important implications for patent claims, formulation strategies and bioavailability6. Screening involves recrystallization of the drug from a variety of solvents under different conditions. It is generally highly desirable to identify and develop the most stable polymorph, bypassing forms that are more soluble but metastable. While the latter offer the promise of better solubility, there are challenges in preventing eventual crystallization to the more stable, less soluble form.
Realistically, it may be impossible to find all existing polymorphs at an early stage. This can result in later complications, including patent litigation and lengthy delays in development time. An extreme example of “the polymorph problem” is the drug ritonavir, where late stage appearance of a new, more stable polymorph compromised original product performance, causing its withdrawal from the market7. In recent years, high throughput screening has facilitated form selection at the early stage. However, this does not replace the need for more detailed investigations of form stability. Furthermore, reliable methods are required to confirm the correct polymorphic form during API synthesis and formulation. There are many techniques used, including optical microscopy, XRD, differential scanning calorimetry (DSC), Fourier Transform infrared (FTIR), near infrared (NIR), Raman, and solid-state nuclear magnetic resonance (ssNMR).
Hydrates/Solvates
In addition to polymorphs, solvated or hydrated drug crystal forms occur. Their crystal structure may or may not be different from the anhydrous form, and they can be stoichiometric or non-stoichiometric in composition. API hydrates (sometimes referred to as pseudopolymorphs) can be isolated and identified through screening studies using organic solvent-water mixtures, but may also form spontaneously in the presence of excess moisture. Conversely, hydrates can lose water upon drying, leading to physical form changes8. Measurement of API water uptake/loss over the entire humidity range is essential, and should be supplemented by DSC, thermogravimetric analysis (TGA), and/or XRD; Raman, and NIR are well suited to monitor hydrate formation during processing, such as in wet granulation. Hydrates with adequate physical stability are suitable for development if the API interactions with water are well understood and can be controlled during formulation and storage.
Traditionally there has been less interest in developing solvates due to safety and regulatory concerns. Nonetheless, a solvate may be considered for development if it can meet essential solubility and bioavailability requirements. For this reason, solvate screens are often conducted in parallel with other solid form screens, and the resulting stable solvates are included in patent claims. Similar to hydrates, solvation and desolvation behavior of the API should be monitored with appropriate analytical methods. Solvate drug products are relatively rare, but are represented by several marketed products, including warfarin sodium and atorvastatin calcium (Lipitor).
Co-crystals
Unlike solvates and hydrates, these materials are designed through crystal engineering, i.e., two crystalline solids interact to form a new crystalline entity. The new structure is stabilized through hydrogen bonding modes, which can be confirmed with various techniques (FTIR, DSC, XRD, X-ray crystallography)9. Co-crystals have generated considerable interest over the last decade for their potential to improve API solubility while maintaining acceptable stability.
As with other drug physical forms, co-crystals screening systems are available, allowing rapid optimization of conditions to produce the desired products.
The ability of compounds to form co-crystals will be significantly influenced by structural factors such as the type and number of substituents. In addition, regulatory perspective on co-crystals continues to evolve. To this end, the FDA issued revised draft guidance in 2016.
Amorphous
As stated above, there is increasing interest in the use of non- crystalline (amorphous) API forms as drug substance and product. There are many ways of preparing amorphous materials, including hot melt extrusion and spray drying10. And while amorphous drugs are attractive for solubility enhancement, they are inherently unstable and pose challenges for formulation due to chemical reactivity and hygroscopicity. Understanding and preventing crystallization of amorphous solid formulations (e.g., through use of polymeric crystallization inhibitors) is key to their successful development. Detection and quantification of crystalline content in an amorphous matrix requires appropriate methods; XRD is a common technique, along with spectroscopic methods (FTIR, NIR, Raman, ssNMR), isothermal microcalorimetry, and DSC.
The glass transition temperature (Tg), which can be measured by DSC, signifies conversion of amorphous to crystalline state, and is used to predict amorphous form stability. In general, the Tg value of amorphous material should be as high as possible to maintain adequate physical stability, although molecular mobility aspects can affect this caveat. It is important to note that Tg decreases significantly in the presence of moisture, leading to crystallization. In some cases, this problem can be minimized by storage of the amorphous material, at low humidity and/or by use of a desiccant.
Stability
Besides solubility, physical and chemical stability of solid drug forms must be adequately characterized. To accomplish this, solids are subjected to stresses (heat, humidity, light) and analyzed. Chemical stability evaluates a drug’s susceptibility to degradation (acid, base, oxidant), and is investigated in solution as well as in solid-state, typically with chromatographic methods. From a physical form standpoint, the possibilities of unwanted crystallization, hygroscopicity, and amorphization (which may occur during milling or micronization) must also be investigated. Furthermore, the stability characteristics of the solid drug influence how it is packaged and stored: desiccant to prevent moisture uptake, amber glass containers to minimize light exposure, refrigeration/freezing to prevent heat-induced degradation, are all considerations, particularly during transport.
Formulation Considerations
During manufacture, an API is exposed to multiple processes on its way to a finished product. In formulations, the drug is combined with a variety of excipients; drug-excipient incompatibility can compromise physical and chemical stability of the API11. A well-known example of chemical incompatibility is the Maillard reaction, where browning occurs due to interaction between amine-containing drugs and reducing sugars such as lactose. Rational choice of excipients is thus critical and should be supported through preformulation experiments – frequently high-performance liquid chromatography (HPLC) and DSC are employed for such screening.
As raw materials, characteristics of the solid drug and associated excipients (e.g., moisture content) may adversely influence tableting processes such as compression or granulation, leasing to physical defects, dissolution problems, and even product failure. Sensitivity of API and excipients to manufacturing conditions should also be considered. Finally, increasingly complex dosage forms require an understanding of how other components can affect API behavior. For example, properties of polymers used in delivery devices (molecular weight, degree of cross-linking, crystallinity) can have a significant effect on drug release.
Case Studies
These studies describe some approaches to solid-state characterization and formulation of poorly soluble APIs.
Compound A
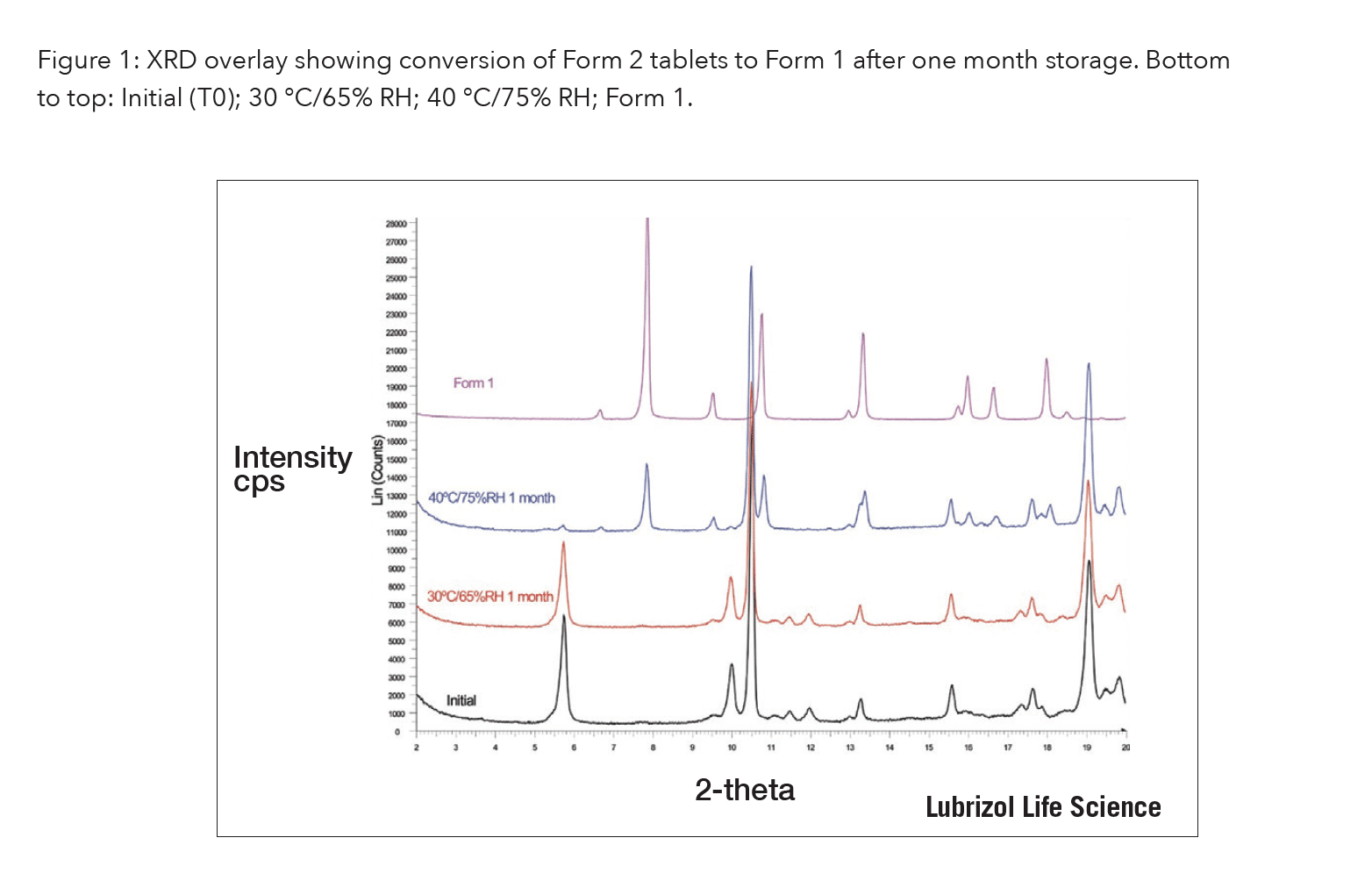
Along with the most stable but least soluble polymorph (Form 1), a metastable polymorph (Form 2) was identified which increased the dissolution rate 3-fold. Form 2 was successfully scaled up and formulated as a tablet. However, during stability studies, Form 2 tablets quickly converted to Form 1 at elevated temperature/high humidity conditions, Fig. 1. In summary, the solubility advantage of the metastable form could not overcome its physical instability.
A second approach to improve API solubility used spray drying to prepare amorphous solid dispersions. Success here was highly dependent on the polymeric excipient used. Certain dispersions remained amorphous by XRD and showed enhanced dissolution compared to the API alone, but others were physically unstable, exhibiting crystallization. Early screening of polymers for compatibility with the API was thus critical for optimizing the amorphous formulation.
Compound B
The starting API was a crystalline acid; one approach to enhance solubility was conversion of the acid to an in situ sodium salt during granulation by addition of sodium hydroxide. An in-process FTIR test was used to confirm conversion, as spectral characteristics of the acid and sodium salt forms are quite different. In the finished tablet product, XRD patterns were utilized to confirm the absence of the acid, which is distinct from that of the salt.
A second approach to enhance solubility involved injection molding. The resulting extrudates, which contained acid API, polymer, and a plasticizer, improved bioavailability and maintained physical stability. Raman spectroscopy confirmed stabilizing interactions of the drug with the polymer in the amorphous matrix.
Fig. 1: XRD overlay showing conversion of Form 2 tablets to Form 1 after one month storage. Bottom to top: Initial (T0); 30 °C/65% RH; 40 °C/75% RH; Form 1.
 Conclusion
Conclusion
Properties of solid drugs impact all stages of drug development, from synthesis to production of clinical and commercial supplies. Appropriate analytical methods are needed not only to generate information during form screening, but also for troubleshooting unexpected problems with solid drug substance and product.
References
- GL Amidon, H Lennermas, VP Shah, and JR Crison, Res. 12, 413 (1995).
- A Avdeef, Absorption and Drug Development, 2nd edition, John Wiley & Sons, Hoboken, NJ (2012).
- S Kumar and P Singh, Pharma Innovation 5, 23 (2016).
- TS Wiedmann and A Naqwi, Asian Pharm. Sci. 11, 722 (2016).
- SR Modi, AJK Dantuluri, SR Perumalla, CC Sun, and AK Bansal, Growth Des. 14, 5283 (2014).
- R Thakuria and TS Thakur, Comprehensive Supramolecular Chemistry II, Elsevier, Oxford, 5, pp. 283-309 (2017).
- J Bauer, S Spanton, R Henry, J Quick, W Dziki, W Porter, and J Morris,. Pharm 18, 859 (2001).
- Y-H Kiang, E Cheung, PW Stephens, and K Nagapudi, Pharm Sci. 103, 2809 (2014).
- S Bhardwaj, M Lipert, and A Bak, Pharm. Sci. 106, 31 (2017).
- M Descamps, Drug Deliv. Rev., 100, 1 (2016).
- R Chadu and S Bhanderi, Pharm. Biomed. Anal. 87, 82 (2014).