Mass Spectrometry in Bioanalysis
Liquid chromatography-tandem mass spectrometry (LC/MS/MS) is the preferred method for the fast and sensitive quantitation of small molecules, peptides, and proteins in complex matrices including plasma, blood, urine, feces, and tissue1. For most compounds, a mass spectrometer is more sensitive and significantly more specific than other LC detectors such as UV-Vis, fluorescence, and refractive index routinely achieving nano- to picogram-per-milliliter detection. LC/MS/MS can analyze compounds that lack a suitable chromophore and can identify components in unresolved chromatographic peaks, reducing the need for ideal chromatographic conditions.
Mass spectrometers use an ion source to generate ions, then sort and identify those ions in the mass analyzer according to their mass-to-charge (m/z) ratios. Different types of ion sources commonly used include, among others, electrospray ionization (ESI) and atmospheric pressure chemical ionization (APCI).
Triple-Quadrupole LC/MS/MS Mass Spectrometers
A quadrupole mass analyzer consists of four parallel rods arranged cubically. Analyte ions are selectively focused down the rods via electromagnetic fields generated by voltages applied to the rods.
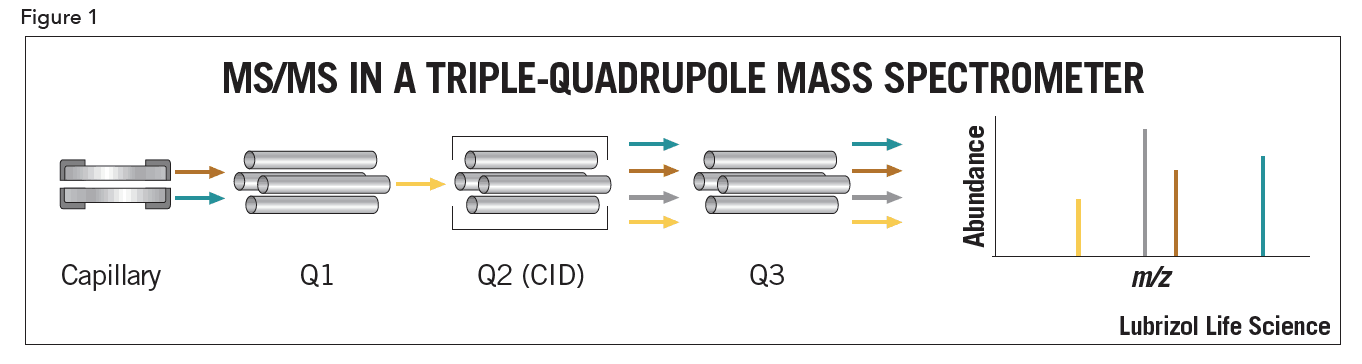
Triple-quadrupole systems (Figure 1) are ideal for quantitative work, as they allow for linear, highly sensitive, and simultaneous multi-component analysis. In triple-quadrupole instruments, the first quadrupole (Q1) is used to select a precursor ion. Collision induced dissociation (CID) takes place in the second quadrupole (Q2), called the collision cell. To obtain specificity or structural information, analyte ions are fragmented by colliding them with neutral molecules. Applied voltages add energy to the analyte ions, increasing collisions which cause fragmentation. The third quadrupole (Q3) generates a spectrum of the resulting product ions. A major advantage of LC/MS/MS is the ability to use Q1 to discard non-analyte ions, resulting in reduced need for sample cleanup and optimum chromatography.
Ionization Techniques
The introduction of atmospheric pressure ionization (API) techniques greatly increased the number of compounds that could be analyzed by LC/MS. In API, the analyte is ionized first at atmospheric pressure then mechanically and electrostatically separated from the neutral molecules. Common API techniques are electrospray ionization (ESI) and atmospheric pressure chemical ionization (APCI).
ESI generates analyte ions in solution before it reaches the mass spectrometer (Figure 2). Because ESI is solution based, mobile phase selection can affect sensitivity, and is an important consideration during method development. The LC eluent is sprayed (nebulized) into a chamber in the presence of a strong electrostatic field and heated dry gas. The electrostatic field causes further dissociation of the analyte while the gas evaporates the solvent. As the droplets shrink, the charge concentration in the droplets increases. Eventually the repulsive forces between ions with like charges exceed the cohesive forces and ions are ejected (desorbed) into the gas phase. These ions are attracted to and pass through an orifice into the mass analyzer.
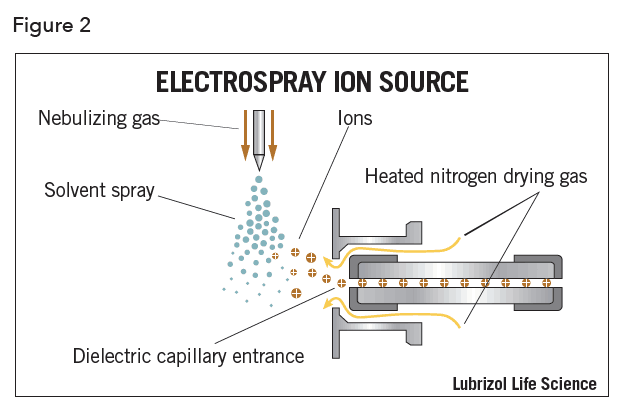
Electrospray ionization has many advantages including its applicability to a wide range of analytes, including high molecular weight biomolecules. This relatively “soft” ionization technique is particularly useful in analyzing thermally labile compounds, which can fragment in the source. The primary disadvantage of ESI is the possibility of ion suppression or enhancement, caused by competition between ions for ejection from the droplet during desolvation.
In APCI, the LC eluent is sprayed through a heated vaporizer typically kept at 250°C – 400°C (Figure 3). The resulting gas-phase solvent molecules are ionized by electrons discharged from a corona needle. The solvent ions transfer charge to the analyte and these ions then pass through an orifice into the mass analyzer.
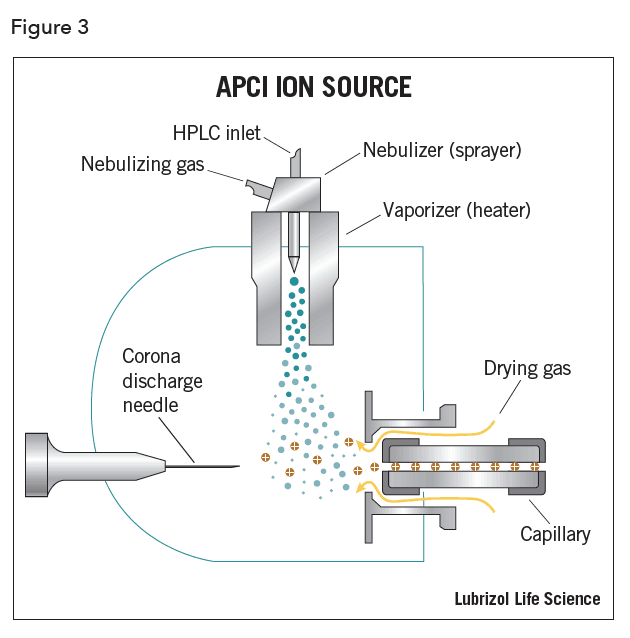
Among the advantages of APCI is the ability to analyze low polarity molecules with a wide dynamic range, usually 4 to 5 orders of magnitude. Higher buffer concentrations can be used, as ion suppression is not typically a concern. However, analytes must be thermally stable and volatile, as ionization is occurring in the gas phase.
Regardless of the type of ionization, voltages applied to the rods focus ions through the optic path of the mass analyzer.
Being highly sensitive and specific, LC/MS/MS allows for reduced chromatographic separation and minimal sample preparation. However, matrix components not removed during sample clean-up can cause a common issue referred to as ion suppression or matrix effects. Ion suppression is not thoroughly understood and is currently an active area of research.2
Ion suppression is the change in response for a given concentration of target analyte in the presence of other sample components. Matrix components in biological fluids such as salts, phospholipids, and metabolites, can cause ion suppression or enhancement of the target analyte response. Typically, ESI is more prone to ion suppression than APCI, and polar compounds are more susceptible than non-polar (hydrophobic) compounds. Since ESI response is governed by droplet size reduction, any component present in the sample that affects solution boiling point or surface tension can potentially be problematic.
The degree of ion suppression can be minimized with different clean-up procedures. The most selective clean-up results in the least amount of ion suppression. Higher suppression in protein precipitation vs. liquid/liquid extraction has been correlated to the higher amount of non-volatile material present in the sample3.
To address these difficulties, stable isotope-labeled internal standards are often used to normalize the effects of ion suppression as they match the ionization properties of the analyte. The internal standard is a compound that closely matches the structure of the analyte of interest. During analysis, it is added in a constant amount to all samples to correct for the loss of analyte during sample preparation, or to correct for ion suppression. Structural analogs of the analyte are generally used if stable isotope internal standards are not available.
Sample preparation
In LC/MS/MS, proper sample preparation is key to success and is focused on concentrating the analyte and removing compounds that can suppress ionization. Common matrices for bioanalytical studies contain different components that need to be removed. Sample preparation is therefore often specific to the sample matrix. For example, blood plasma major constituents include proteins, sugars, salts, lipids, as well as numerous peptides and small molecules. Urine is a less complex matrix, comprised primarily of salts and urea.
Sample preparation has three goals: removal of protein-related materials, elimination of endogenous compounds, such as phospholipids which can cause ion suppression or enhancement, and concentration to increase assay sensitivity.
Common sample preparation techniques are protein precipitation, solid phase extraction (SPE), and liquid/liquid extraction (LLE).
Protein precipitation is performed by simply adding a large proportion of acetonitrile to the sample, usually a ratio of three to four volumes of acetonitrile to plasma. A lower ratio can result in insufficient protein precipitation, and a larger ratio may over-dilute the sample. Protein precipitation however, only addresses the removal of proteins. Phospholipids and other matrix contaminants that remain may cause ion suppression or enhancement, resulting in inconsistencies and inaccuracies in detection and quantitation of the analyte. Despite these shortcomings, precipitation remains a popular method for sample clean-up because it is relatively fast and inexpensive.

In contrast to protein precipitation, LLE creates cleaner extracts, but the procedure is cumbersome and has many drawbacks. To transfer an ionizable analyte into the organic extraction solvent, it first needs to be converted to a non-ionic form in its aqueous medium using either high or low pH. It should be noted that this procedure is not suitable for zwitterions. Next, an appropriate solvent needs to be found that efficiently and preferentially extracts the analyte. Multiple extraction steps are often needed, as a single step rarely extracts the analyte quantitatively. Reproducibility must be maintained with an internal standard that has extraction properties very similar to those of the analyte.
LLE has advantages over protein precipitation because the sample can be concentrated. However, selecting the right solvents for analyte extraction makes development labor intensive. Other factors to be considered are solvent cost and disposal, as well as the lack of potential for automation.
SPE solves problems associated with both protein precipitation and LLE. It is amenable to small sample volumes and can be easily automated. SPE is usually carried out off-line using single cartridges or 96-well plates and has the advantage that specific protocols can be developed, allowing very selective sample cleanup. The problem of ion suppression can be solved using an appropriate solid-phase extraction method.
SPE offers many stationary phases for sample clean-up, the most common being normal-phase, reversed-phase, and ion exchange solid phase extraction. Using the correct sorbent for removal of endogenous components results in a very clean sample for subsequent analysis.
Normal phase SPE procedures typically involve a polar analyte, a mid-to-nonpolar matrix (e.g. acetone, chlorinated solvents, and hexane), and a polar stationary phase. Retention of the analyte is primarily due to hydrophilic interactions such as polar-polar interactions between the analyte and stationary phase, hydrogen bonding, or dipole-dipole interactions. A compound adsorbed by these mechanisms is eluted by using a solvent that disrupts the binding mechanism, usually a solvent more polar than the sample’s original matrix.
Reversed-phase has become the prevalent mode of SPE because of the diversity of aqueous samples such as environmental water, fruits and vegetables, beverages, and biological fluids. Reversed-phase SPE involves a polar, usually aqueous, or moderately polar sample matrix, and a nonpolar stationary phase. The analyte of interest is typically mid-to-nonpolar. Retention of the analyte is due primarily to hydrophobic interactions such as van der Waals forces. Elution of the analyte is accomplished using a nonpolar solvent to disrupt the forces that bind the compound to the packing.
Ion exchange SPE separates analytes based on electrostatic interactions between the analyte and positively charged groups on the stationary phase. Anion exchange sorbents contain positively charged functional groups that retain negatively charges anions, such as acids. Cation exchange sorbents contain functional groups that retain cations, such as bases. For ion exchange to occur, the stationary phase and sample must be at a pH where both are charged. A solution having a pH that neutralizes either the compound’s functional group or the functional group on the sorbent surface is used to elute the compound of interest.
Conclusions
Using LC/MS/MS for bioanalysis compared to HPLC-UV provides superior sensitivity, lower detection limits and greater specificity, leading to improved data and more confidence in the results. LC/MS/MS significantly reduces sample preparation and LC run times, increasing sample throughput. With the increase in demand for high quality bioanalytical data, it is easy to understand why LC/MS/MS has become the method of choice for quantitative analysis.
References
- Prentis et al. Br. J. Clin. Pharmacology, 1988; 25; 387- 96.
- Topics in Solid-Phase Extraction; Waters Corporation; Technical Bulletin; 2005.
- King, R., Bonfoglio, R., Fernandez- Metzler, C., Miller-Stein, C., and Olah, T.J., Am Soc Mass Spectrom, 2000, 11, 942-951.