Combination Devices
Combination products are defined in 21 CFR 3.2(e) as therapeutic and diagnostic products comprising two or more regulated components (i.e., drug/device, biologic/device, drug/biologic, or drug/ device/biologic) that are physically, chemically, or otherwise combined or mixed and produced as a single entity. Development of combination devices (products that combine a medical device with an active pharmaceutical ingredient (API)) has been a growth area since it was first demonstrated that drugs could be delivered with controlled release kinetics from a polymer.
Combination devices can be designed for both local and systemic dosing. Their potential benefits include reduced side effects from lower localized doses, protection of the drug against in vivo degradation, consistent local and/or systemic drug levels, synergy of therapeutic functions between a device’s structural effects and an API’s chemical effects, increased patient compliance, and delivery of drugs to areas that cannot otherwise be adequately accessed by systemic dosing. Drug/device combinations also offer drug-developers an attractive lifecycle management option.
Types of Combination Devices and Production Processes
Combination devices may be applied topically (ex. patches containing testosterone, nicotine, or contraceptives), mucosally (ex. contraceptive or anti-retroviral vaginal rings, contact lenses, and catheters), implanted in the patient (ex. arterial stents, subcutaneous contraceptive rods, and ocular implants), or somewhere in between (ex. drug-eluting biodegradable sutures). The drugs they contain may be either impregnated or surface coated.
The API(s) can be mixed into a thermoplastic polymer by hot-melt extrusion to yield a uniformly mixed ribbon. The finished device may simply be the appropriately cut ribbon, or the ribbon may be further processed, for example by injection molding. Polymers that have been used to manufacture this type of combination device include polyolefins, polyesters (especially those based on lactic acid, glycolic acid, and caprolactone) and ethylene-co-vinylacetate polymers. Silicone rubber devices are made by reactive injection molding.
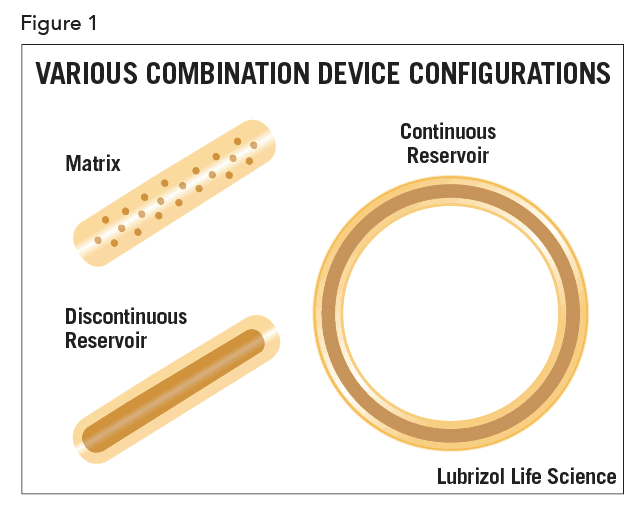
A device developer may choose to disperse the API in the device uniformly or in a spatially-localized way. If the concentration of the API is uniformly distributed in the device, the device is called matrix-type. In other cases, the API is localized in a section of the device, as illustrated schematically in Figure 1 for various configurations.
Lastly, the drug may simply be coated on top of the device as is done with urethral catheters where antimicrobials are applied to prevent pathogenic colonization of the catheter.
Biocompatibility
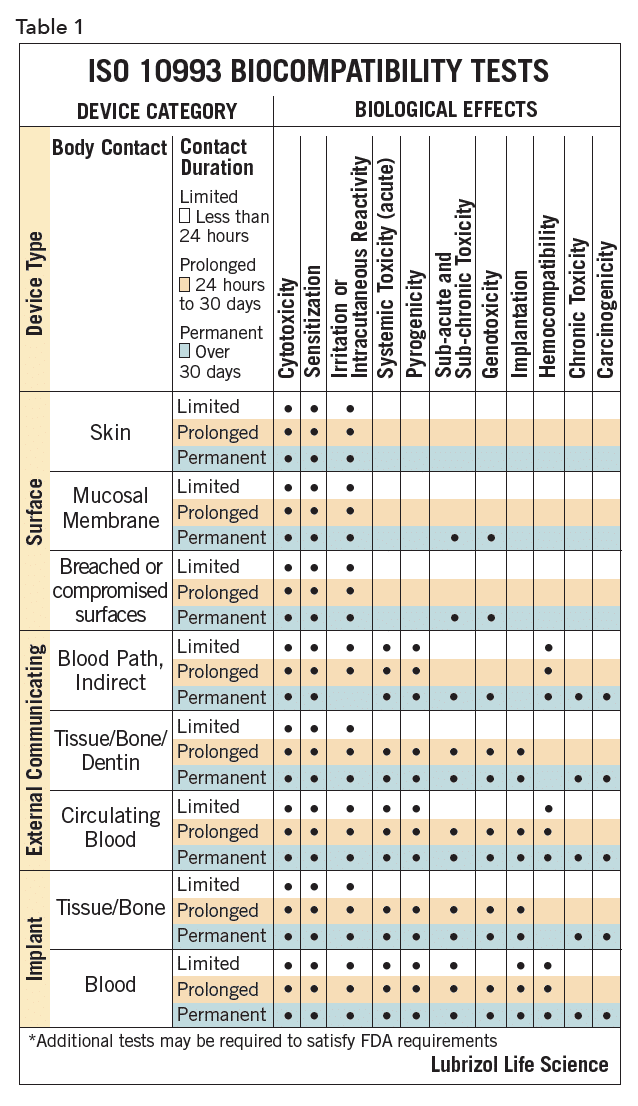
The biocompatibility of device polymers is determined by several tests, as outlined in ISO 10993 and USP <1031>, designed to document that the final product, when used as indicated, will be safe. The specific tests for a given device are determined by the intended point of contact and duration of use for that product. These ISO and USP documents provide guidance on the needed testing for a given contact/duration scenario. For example, polymers for devices for >30 days mucosal contact would be classified as USP Class VI or ISO surface/mucosal/permanent. These designations dictate the suite of biocompatibility tests needed, as summarized in Table 11. Manufacturers of polymers for devices are generally cautious, and device developers should ensure that their chosen polymer, as with any key component, will be available at the appropriate point in the commercialization process.
Biocompatibility is perhaps the most significant regulatory issue in device development, especially for devices that are designed for long-term indwelling application such as stents, some catheters, and slow-release devices. There are several well-known biocompatible materials, including the polymers which were listed earlier, ceramics and other inert metals such as stainless steel and certain chrome alloys. Biocompatibility is often determined by implanting the de- vice into animals, or administering extracts from devices to animals, followed by systemic and histological analysis.
Phases of Combination Device Development
The development of a combination device follows stages similar to that of a conventional drug product, i.e., development of analytical methods, chemical compatibility of device components and drug, formulation development, stability testing, drug release testing, reproducibility of manufacture, in vivo toxicology, followed by human clinical trials phase I to III and finally FDA approval and commercialization. Approval of combination devices is more involved than other drug delivery types, as several regulatory divisions within the same agency may be involved. From the FDA’s website, “FDA expects to receive large numbers of combination products for review as technological advances continue to merge product types and blur the historical lines of separation between FDA’s medical product centers, which are made up of the Center for Biologics Evaluation and Research (CBER), the Center for Drug Evaluation and Research (CDER), and the Center for Devices and Radiological Health (CDRH). Because combination products involve components that would normally be regulated under different types of regulatory authorities, and frequently by different FDA Centers, they raise challenging regulatory, policy, and review management challenges. Differences in regulatory pathways for each component can impact the regulatory processes for all aspects of product development and management, including preclinical testing, clinical investigation, marketing applications, manufacturing and quality control, adverse event reporting, promotion and advertising, and post-approval modifications”.2
In preclinical development, issues of drug/polymer/process compatibility, biocompatibility, mechanical properties and drug release are addressed.
Initially, the stability of the API in the polymer under processing conditions is ascertained. Since device manufacture often involves elevated temperatures, the thermal stability of the API in the polymer is of interest and must be determined early.
Where the mechanical properties of the devices are important for specific biological function or durability, these are optimized within a usage model framework. For example, stent designs are modeled and tested in a way that simulates arterial insertion and in situ expansion with an angioplasty balloon and then tested in simulated arteries for their durability and stability. Intravaginal rings have physical properties that allow their insertion by the user while ensuring that the device remains comfortably in place once fitted.
Controlled drug release is often the only functional purpose of a combination device, as with patches, inserts, some implants, and intravaginal rings. In these cases, optimizing drug release kinetics while maintaining device safety is of paramount importance. Drugs may be released with zero-order, first-order, or pulsatile kinetics as shown schematically in Figure 2.
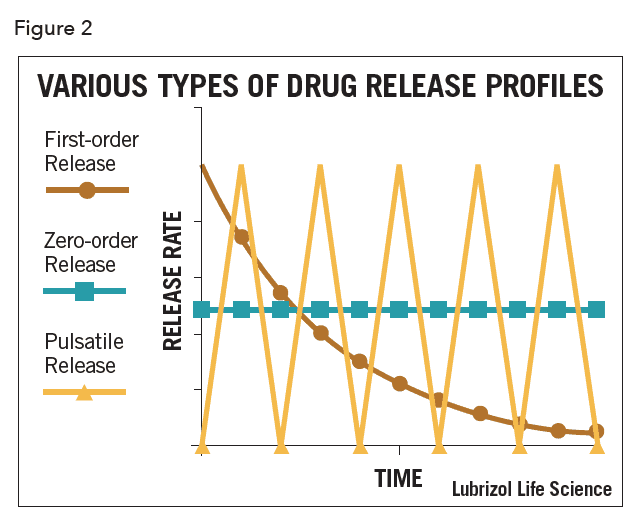 Most commonly, and especially for matrix-type devices, drug release follows first-order kinetics, often with an initial burst of drug. In practice, a zero-order profile is often desired, though considerable design effort and perhaps even complex manufacturing processes are required to achieve it. In the case of vaginal rings and similar drug-filled monolithic devices, zero-order release has been achieved by localizing the API in a reservoir within the device (as shown in Figure 1). This can be achieved by filling a cavity within the device with an API formulation, or by coating a matrix device with pure polymer by spraying, dipping, or co-extrusion. Recently, a technology called Microreservoir™ was developed that incorporates the API into micro-capsules before mixing with the polymer. The drug release kinetics are closer to zero-order, especially over extended periods.
Most commonly, and especially for matrix-type devices, drug release follows first-order kinetics, often with an initial burst of drug. In practice, a zero-order profile is often desired, though considerable design effort and perhaps even complex manufacturing processes are required to achieve it. In the case of vaginal rings and similar drug-filled monolithic devices, zero-order release has been achieved by localizing the API in a reservoir within the device (as shown in Figure 1). This can be achieved by filling a cavity within the device with an API formulation, or by coating a matrix device with pure polymer by spraying, dipping, or co-extrusion. Recently, a technology called Microreservoir™ was developed that incorporates the API into micro-capsules before mixing with the polymer. The drug release kinetics are closer to zero-order, especially over extended periods.
To determine in vitro release kinetics, a combination device is immersed in a fluid which is replaced daily to maintain sink conditions. The fluid should simulate in vivo conditions in some respect, and if the device is designed to slowly dissolve in vivo, it should do so in this fluid. If the device is not intended to dissolve in vivo the fluid should not be a solvent for the polymer. The amount of drug released from the device in samples of medium removed periodically is determined, usually by HPLC. In vivo release kinetics are similarly determined by measuring levels of the drug in tissue or biological fluid over a chosen period.
The stability of devices is tested in similar ways to other drug formulations. Using ICH guidelines, the devices are stored at controlled temperature and humidity, preferably in the primary packaging chosen for the product, removed at set timepoints and their quality-determining properties measured. Typically,the API and related substances are assayed to ensure no drug-degradation and then drug release, physical properties, and appearance are measured. For GLP toxicology and GMP human clinical trials, a stability study would also be conducted using devices made for the study to demonstrate that all devices had comparable properties no matter what point in the study they were used. This can be especially important in long trials.
Conclusions
A wide range of combination devices have been commercialized and continue to be developed. The regulatory pathway can be challenging but is manageable and can even present some opportunities for developers. Using an experienced development partner can mitigate much of the risk and shorten the time between developmental inflection points. The added value derived from using passive devices to deliver drugs in a controlled way, or control the body’s response and reaction to an active device by incorporating response-modulating drugs within the device, ensures that this field will continue to see growth.
References
- ISO 10993-1, 3rd Edition 2003-08-01 “Biological Evaluation of Medical Devices”- Part 1: Evaluation and Testing
- https://www.fda.gov/combination-products/about-combination-products