Aseptic Manufacturing and Sterile Fill-Finish for Complex Drug Products
Key Points
- Many complex drug products are not amenable to terminal sterilization, leading to increased demand for aseptic manufacturing and sterile fill-finish capabilities.
- Aseptic processing is uniquely challenging because it requires careful planning, thoroughly trained personnel, and specialized facilities/equipment to properly execute.
- Cleanroom facilities and aseptic processes are designed to minimize contamination risk from personnel, materials, and equipment.
- Sterile lyophilization requires investment into specialized equipment, facilities, and knowledge, so choosing the right CDMO to scale-up your process is critical for successs.
When is Sterile Fill-Finish Required?
Drug products that are delivered via the parenteral, ophthalmic, inhaled, or otic route present an increased risk of infection or harm because they bypass many of the body’s natural defenses. To ensure patient safety, the FDA requires that drug products delivered via these routes be supplied as sterile products. This designation includes many complex drug products, including ophthalmic suspensions, sterile injectables, reconstituted lyophilized powders for injection, and aqueous-based aerosols for inhalation.

Figure 1: Global market for contract manufacturing of sterile injectables and infusions
As these complex APIs and formulations become more common, there is an increased need for aseptic operations, much of which is being addressed by contract manufacturers (Figure 1). In general, there are two ways to manufacture a sterile drug product:
- Terminal Sterilization: A process that involves filling and sealing product containers under high-quality environmental conditions, then subjecting the product in its final container to a sterilization process such as heat or irradiation.
- Aseptic Manufacturing and Sterile Fill-Finish: A process in which the drug product, container, and closure are first subjected to sterilization methods separately, as appropriate, and then brought together (aseptic manufacturing). Because there is no process to sterilize the product in its final container, it is critical that containers be filled and sealed in an extremely controlled environment (sterile fill-finish). This represents one of the hardest challenges in pharmaceutical manufacturing.
The FDA has made it clear in multiple guidances that aseptic manufacturing and sterile fill-finish should only be employed when terminal sterilization is not feasible because aseptic processing involves more variables and therefore carries more risk. However, as formulations become more complex, a growing number of drug products and containers cannot be terminally sterilized due to degradation or loss of performance when exposed to heat or radiation.
When terminal sterilization is not possible, manufacturers of parenteral, ophthalmic, inhaled, and otic drug products must turn to aseptic manufacturing and sterile fill-finish.
Why is Aseptic Processing Challenging?
Aseptic processing is uniquely challenging because it requires careful planning, thoroughly trained personnel with the appropriate mindset, and specialized facilities/equipment/processes to properly execute. Ensuring sterility is not a trivial task, and failure can have catastrophic—even life-threatening—consequences for a patient. Ultimately, the goal of an aseptic manufacturing process is to completely eliminate opportunities for contamination, whether it be from microorganisms or particulates that could harm a patient when administered. Any of the following can be sources of contamination in an aseptic processing and sterile fill-finish operation:
- Personnel
- Drug Product Components and Container Systems
- Cleanroom Facilities
- Equipment and Processes
The success of aseptic processing and sterile fill-finish operations relies on mitigating contamination from each of these sources.
Considerations for Aseptic Processing and Sterile Fill-Finish
Drug Product Components
When the term “sterile fill-finish” is used, often the first drug products that come to mind are sterile injectables (e.g., liquids filled in vials or syringes). While liquid solutions, suspensions, and emulsions are common candidates for sterile fill-finish operations, powder fills and lyophilization (i.e., freeze-drying to preserve and obtain greater stability) are also performed under aseptic conditions.
Regardless of the form factor, each individual component comprising a drug product must be sterilized prior to aseptic fill-finish. This includes active pharmaceutical ingredients (APIs), water for injection (WFI), and any other excipients that are part of a formulation. Equipment and container systems must also be sterilized. There are several FDA-recommended methods to sterilize components/equipment:
- Heat Sterilization: The most widely used sterilization method for sterilizing processing equipment, wherein a component is exposed to dry heat or moist heat (autoclaving).
- Radiation Sterilization: A method wherein a component is exposed to electromagnetic radiation (e.g., gamma rays) to kill microorganisms. May be useful if a component is heat-sensitive.
- Filter Sterilization: A method wherein a component is dissolved in solution (WFI or another suitable vehicle) and passed through a membrane with a 0.2 micron or smaller pore size. This removes microorganisms such as bacteria but cannot be used if the particle size in a formulation component is too large (e.g., suspensions). Also, this does not remove bacterial endotoxins which must be controlled at the raw material stage.
- Ethylene Oxide Gas Sterilization: A method used mainly to sterilize bulk API powder and container systems wherein a component is exposed to ethylene oxide (EtO) over an extended period of time. This method is used to sterilize heat- and moisture-sensitive components.
Selection of a sterilization method should involve studies that ensure the process is appropriate for a given component and does not cause degradation or failure. Each of these methods must also be accompanied by written procedures and appropriate specifications for acceptance or rejection of contaminated components.
Container System
Aseptic processing and sterile fill-finish operations can be applied to a range of container systems made of either glass or plastic, including vials, syringes, bottles, cartridges, and ampoules. For ophthalmic preparations, single dose blow-fill-seal (BFS) containers molded from plastic and newer multidose, sterile-delivery packaging may also be options.
Glass containers typically undergo a pre-sterilization process to remove foreign matter. The process consists of a series of wash and rinse cycles in high purity water (WFI if the container is for a parenteral drug product). The containers are then typically subjected to dry heat for sterilization and depyrogenation to destroy bacteria and remove endotoxins. The exact parameters of sterilization and depyrogenation are based on validation studies which vary conditions and measure the uniformity of sterilization and depyrogenation under different container loading conditions.
Plastic containers are heat sensitive, so they are typically sterilized via radiation, sterilizing gas, or other means not involving heat. EtO is an example of an effective sterilizing gas that is often used. If a sterilizing agent like EtO is employed, residuals from the agent should be measured and kept within regulatory limits.
Cleanroom Facilities
Aseptic processing and sterile fill-finish operations take place in cleanrooms that are designed to accommodate the flow of personnel, materials, and equipment during a manufacturing process. A cleanroom is a controlled environment that defines personnel access, levels of contamination, pressurization, and temperature/humidity. The cleanroom suite comprises multiple rooms with varying levels of control. This is referred to as a “cascading” design (Figure 2), wherein the level of air quality increases as you move closer to the aseptic core of the facility (a Class 100 space). The flow of personnel, materials, and equipment starts in supporting areas where gowning, nonsterile preparation, and formulation activities can take place.
The highest level of air quality is maintained in the Critical Area, which has an ISO 5 certification (Class 100, or Grade A). Activities conducted in the Critical Area include manipulations (e.g., aseptic connections, sterile ingredient additions) of sterile materials prior to and during filling and closing operations. Because the drug and components are exposed to the environment, it is essential that the personnel’s actions, amount of contaminants, pressurization, temperature, and humidty in this space are stringently controlled, constantly maintained, and continuously monitored.

Figure 2: Pharmaceutical cleanroom design
Air quality is maintained via specialized heating, ventilation, and air conditioning systems. These are complex systems that engage High Efficiency Particulate Air (HEPA) filters supplying ISO 5 air in a unidirectional, laminar flow to sweep particles away from the fill-finish area and minimize contamination potential. Critical Areas must be designed to minimize turbulence and stagnant air, which requires in-depth studies of airflow and the sweeping action of the laminar flow.
Environmental Monitoring (EM) activities are vital to the operation of a cleanroom. Proper EM requires qualified methods to regularly collect, evaluate, and interpret data on the air, surfaces, and personnel in the cleanrooms. Detection of microorganisms is achieved by the following methods:
- Personnel Monitoring: A process that involves sampling an operator’s gloved hands immediately after performing critical interventions as well as the entire sterile gown prior to existing the sterile suite. This is performed with touch plates which are analyzed by a quality control lab for viable microorganisms.
- Non Viable Particulate Monitoring: A process that involves sampling the air for a quantity of micron-sized particulates per cubic meter of air. This is performed constantly at critical sites and routinely at noncritical sites via sophisticated equipment utilizing validated processes.
- Surface monitoring: A process that involves sampling contact surfaces, floors, walls, and equipment for viable organisms on a regular basis. This can be performed with touch plates, swabs, and contact plates.
- Active air monitoring: A process that utilizes devices that regularly sample the air for viable organisms, including impaction, centrifugal, and membrane samplers.
- Passive air monitoring: A process that utilizes collection devices such as settling plates (petri dishes containing nutrient growth medium exposed to the environment) that are analyzed by a quality control lab for viable microorganisms.
EM should allow a manufacturing organization to quickly recognize trends and identify sources of contamination, enabling corrective action before product contamination occurs. According to FDA guidance, written SOPs for an environmental monitoring system should address frequency of sampling, timing of sampling, duration of sampling, sample size, specific sampling equipment and techniques, alert and action levels, and appropriate response to deviations from alert or action levels.
Equipment and Process
Sterile fill-finish operations can take several forms, including hand-filling of clinical trial material (CTM), fully automated high-speed filling lines, and sterile lyophilization to name a few. In this section, we will focus on solution filling of glass vials and syringes, which is performed with semi-automated or fully-automated fillers (Figure 3).

Figure 3: An operator interacts with a filler equipped with a restricted access barrier system (RABS)
Process design for aseptic manufacturing focuses on minimizing exposure of sterile items to contamination hazards. This means that processes should flow in a logical manner and equipment should be arranged in a way that minimizes unnecessary activities and movement by personnel. Because interventions by personnel can increase the risk of contamination, sterile fill-finish equipment is often designed to minimize the need for human interaction. Many vial and syringe fillers come equipped with in-line weight checking, allowing operators to monitor the weight of products without contacting the drug product. Fillers may also have automated rejection and vision systems to sort and process vials and syringes as they are filled. Sterilize-In-Place (SIP) technology allows for sterilization of equipment without complex manipulations and aseptic connections between process components.
A Key Step: Aseptic Process Simulations (APS) or Media Fills
The aseptic qualification of a process within an area is called an Aseptic Process Simulation (APS) or Media Fill. During the development of the APS, all known inherent and noninherent interventions and activities are identified. Each intervention and activity is rated under protocol in a risk assessment. The interventions are categorized as minor, major, or critical during the risk assessment.
From that determination, all major and critical interventions are further studied in an airflow visualization study (smoke study) to understand the effects of the interventions on the product. Furthermore, all interventions and activities are studied in an Environmental Monitoring Process Qualification (EMPQ) and a Media Fill. During the EMPQ, the controlled rooms are studied under protocol for viable and nonviable recovery during periods of rest and while in full use. Finally, all interventions and activities are performed during a media fill.
During the media fill, a growth medium such as tryptic soy broth is used in lieu of product within the filling process while performing all activities and interventions. At the end of the media fill, the final containers filled with growth media are incubated for multiple weeks and at multiple temperatures to encourage the growth of organisms. At the end of the incubation, each final container is visually inspected for growth.
Learn more about Aseptic Process Simulations and Media Fills in our recent webinar.
The use of barrier systems can further protect sterile products from contamination. The strictest example of this is the use of aseptic processing isolators, which separate the materials inside them from the external cleanroom environment and remove exposure of the sterile product to personnel. Operators manipulate items in the isolator via isolator gloves or half-suits that maintain the barrier with the external environment. Fillers may also be equipped with Restricted Access Barrier Systems (RABS), which also provide a physical barrier to the outside environment and utilize RABS glove ports for interaction with products in the filler. RABS systems are appealing due to their reduced capital investment and start-up time compared to aseptic processing isolators.
Personnel
Any personnel who enter an aseptic manufacturing area must be thoroughly trained in cleanroom procedures and aseptic behavior. While aseptic processing and sterile fill-finish operations are designed to limit human interventions, the actions of personnel in the cleanroom go a long way towards ensuring product sterility. According to the FDA, personnel are a potentially major source of contamination and a proper training program should cover, at a minimum:
- Aseptic technique
- Cleanroom behavior (e.g., move slowly and deliberately, keep all body parts out of the path of HEPA-filtered air between the HEPA filter and the product contact parts)
- Microbiology
- Hygiene
- Gowning
- Patient safety hazards posed by nonsterile drug products
- Specific written procedures covering aseptic manufacturing area operations
Ongoing training and evaluations of cleanroom personnel and procedures are necessary to ensure products are not exposed to contamination risks. Many of the same principles apply to laboratory personnel who are testing aseptic samples and generating microbiological data from the cleanrooms, as they must also avoid contaminating the samples.
Finally, the environmental monitoring team plays a critical role in detecting anomalies or adverse trends in aseptic manufacturing. As stated above, the EM team designs sampling strategies and outlines clear alert and action levels for measurements of contamination. Their vigilance and prompt response to EM test results is vital to maintaining a sterile environment for drug product manufacturing.
Sterile Lyophilization of Parenteral Drug Products
As complex drug products and large molecule formulations become more common, an increasing number of pharmaceutical formulations face stability issues in solution and a ready-to-use liquid dosage form is not possible. To solve this issue, many parenteral drug products undergo sterile lyophilization (i.e., freeze-drying) to generate a stable powder for storage and transport. This is not a new process, and multiple blockbuster drugs (including biologics such as Enbrel, Herceptin, and Rituxan) are possible due to sterile lyophilization. From 2013-2015, half of all new injectable/infusible drug approvals were lyophilized, and market projections estimate this growth will continue in the future (Figure 4).
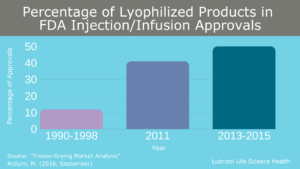
Figure 4: Percentage of lyophilized products in FDA sterile injectable and infusion approvals
The Lyophilization Process
If a product requires lyophilization, vials of solution are partially stoppered during sterile fill-finish transferred to the lyophilizer (Figure 5). Facilities for aseptic manufacturing and sterile fill-finish should be designed such that any transition from filling to lyophilization takes place in a Critical Area with ISO 5 (Class 100) certification. Procedures for transferring product should also be designed to minimize movement of product and reduce risk of contamination. Once inside the lyophilizer, solutions undergo three steps:
- Freezing: The temperature of the solution is lowered at a predetermined rate to ensure complete freezing and a favorable crystal structure in the frozen solid.
- Primary Drying: The pressure inside the drying chamber is gradually lowered to promote drying via sublimation.
- Secondary Drying: The temperature inside the drying chamber is slowly raised under low pressure to drive off any residual solvent that is still chemically bound to the material. After this step, the vials are fully stoppered to minimize further exposure to the outside environment.
These steps require extensive cycle development and customization for each product, and the process can take anywhere from hours to days to complete.

Figure 5: An operator loads vials into a sterile lyophilizer
While lyophilization is an accepted, beneficial technology, it also comes with many challenges:
- Specialized Knowledge Required: Lyophilization cycle development and scale-up rely on a thorough understanding of the freezing and sublimation processes. There are numerous studies that must be performed to understand factors such as crystal structure changes during freezing, heat transfer through a vial, and phase transition temperatures of a product. A full understanding of these product characteristics helps determine the optimal freezing rate and temperature ramping rate in a lyophilization cycle. However, these rates vary for different vials sizes, strengths, and batch sizes of formulations, requiring further investigation.
- Additional Contamination Risk: Transportation and loading of partially stoppered vials into a sterile freeze-drier leaves product exposed to the environment and increases contamination risk. Therefore, all activities associated with lyophilization must be performed in a Critical Area. According to the FDA, the sterilization of lyophilizer is one of the more frequently encountered problems noted during inspections. As a result, many sterile lyophilizers are now equipped with both Clean-In-Place (CIP) and Steam-In-Place (SIP) capabilities. Extensive validation and monitoring of cleaning procedures is required in any lyophilization operation.
- High Capital Investment: R&D development of a lyophilization cycle can take place with pilot scale equipment in small batches. However, large-scale lyophilizers and the associated cleanroom facilities to accommodate sterile fill-finish cost millions of dollars to install and maintain. As a result, both small and large pharmaceutical companies will often transfer their lyophilization processes to CDMOs for clinical and commercial manufacturing. These CDMOs have the equipment and personnel in place to scale-up sterile lyophilization processes.
Choosing the Right Partner for Aseptic Manufacturing and Sterile Fill-Finish
The need for reliable aseptic processing and sterile fill-finish operations will continue to grow as more complex parenteral, ophthalmic, inhaled aqueous aerosol, and otic drug products come to market. And because of the highly specialized nature of these operations, finding the right partner is not always straightforward. Few CDMOs are suitably equipped to handle aseptic processing and sterile fill-finish operations on a clinical or commercial scale.
As we’ve explored in this post, manufacturing of sterile drug products requires purpose-built infrastructure, highly specialized staff, and a commitment to quality. Circumnavigating the challenging process considerations and regulatory requirements of operations such as sterile lyophilization are not trivial tasks, and choosing the right partner to take a sterile product into clinical or commercial production is critical for success.
At Particle Sciences, we are leading the way in commercial aseptic manufacturing and sterile fill-finish of complex drug products, leveraging our decades of know-how as a leading product developer and clinical-stage manufacturer. Our commercial facility is integrated into our existing development and clinical trial manufacturing site, offering customers a seamless flow from development through manufacturing—a one-stop-shop.
Our facility contains ISO 5 Filling Suites for both clinical and commercial production. This includes our Grade A RABS aseptic filling suite, which can process vials, ophthalmic bottles, syringes, and cartridges. Our 120 ft2 lyophilizer builds upon our existing R&D and clinical lyophilization capability (2 R&D and 2 small-scale clinical lyophilizers) and is supported with both Clean-In-Place and Steam-In-Place capabilities for aseptic processing. Along with our sterile fill-finish and lyophilization capabilities, we can also perform particle size reduction and complex formulation activities under aseptic conditions. Finally, our analytical and quality control team works closely with our development and manufacturing staff to ensure your product is manufactured to the highest standards.
If you want to bring a complex drug product to market, look no further than the experts at Particle Sciences. Take the next step and contact us today!
Author: