Protein Structure: Primary, Secondary, Tertiary, Quatemary Structures
Increasingly, drug developers are looking to large molecules, particularly proteins, as a therapeutic option. Formulation of a protein drug product can be quite a challenge, and without a good understanding of the nature of protein structure and the conformational characteristics of the specific protein being formulated, the results can be ruinous. This technical brief aims to give the reader a quick overview of protein structure. It will also cover briefly how protein structure can be affected during formulation and some of the analytical methods which can be used both to determine the structure and analyze the stability of the protein.
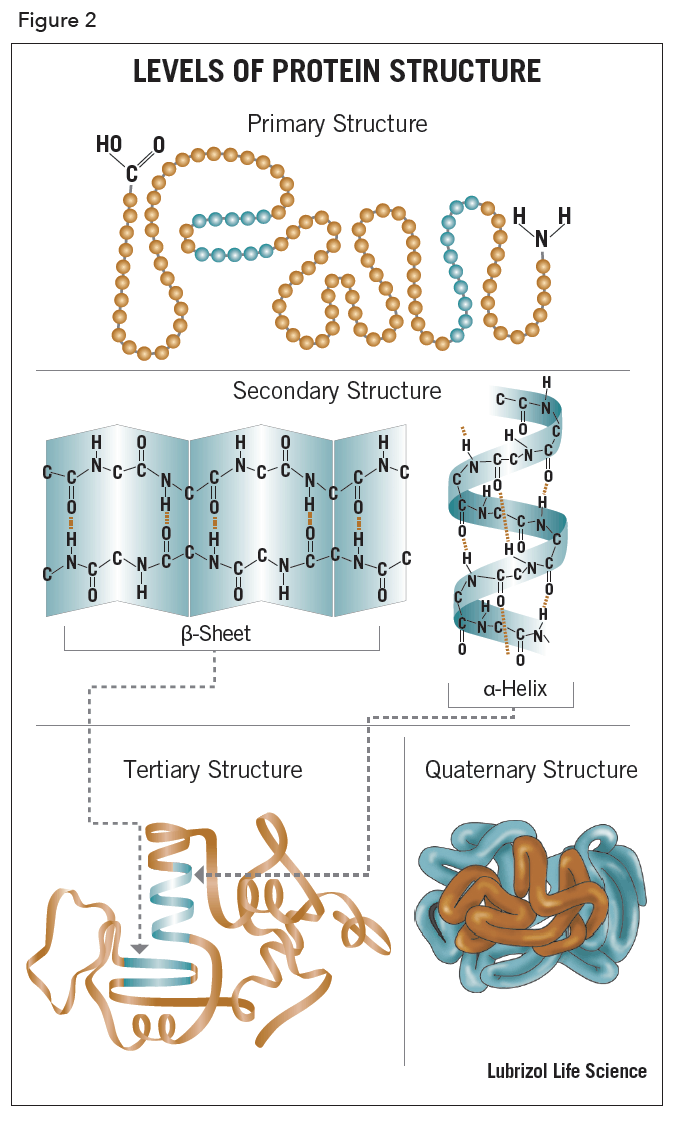
The term, structure, when used in relation to proteins, takes on a much more complex meaning than it does for small molecules. Proteins are macromolecules and have four different levels of structure – primary, secondary, tertiary and quaternary.
Primary Structure
There are 20 different standard L-α-amino acids used by cells for protein construction. Amino acids, as their name indicates, contain both a basic amino group and an acidic carboxyl group. This difunctionality allows the individual amino acids to join in long chains by forming peptide bonds: amide bonds between the -NH2 of one amino acid and the -COOH of another. Sequences with fewer than 50 amino acids are generally referred to as peptides, while the terms, protein and polypeptide, are used for longer sequences. A protein can be made up of one or more polypeptide molecules. The end of the peptide or protein sequence with a free carboxyl group is called the carboxy-terminus or C-terminus. The terms, amino-terminus and N-terminus, describe the end of the sequence with a free α-amino group.
The amino acids differ in structure by the substituent on their side chains. These side chains confer different chemical, physical, and structural properties to the final peptide or protein. The structures of the 20 amino acids commonly found in proteins are shown in Figure 1. Each amino acid has both a one-letter and three-letter abbreviation. These abbreviations are commonly used to simplify the written sequence of a peptide or protein.
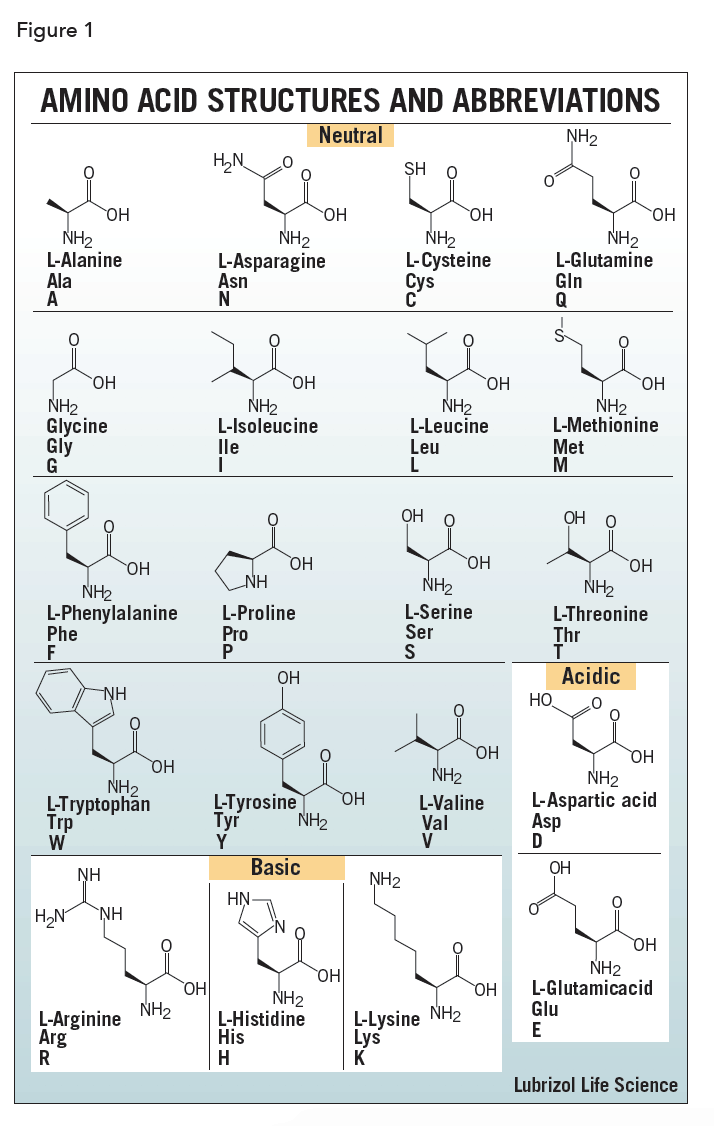
Depending on the side-chain substituent, an amino acid can be classified as being acidic, basic or neutral. Although 20 amino acids are required for synthesis of various proteins found in humans, we can synthesize only ten. The remaining 10 are called essential amino acids and must be obtained in the diet.
The amino acid sequence of a protein is encoded in DNA. Proteins are synthesized by a series of steps called transcription (the use of a DNA strand to make a complimentary messenger RNA strand – mRNA) and translation (the mRNA sequence is used as a template to guide the synthesis of the chain of amino acids which make up the protein). Often, post-translational modifications, such as glycosylation or phosphorylation, occur which are necessary for the biological function of the protein. While the amino acid sequence makes up the primary structure of the protein, the chemical/biological properties of the protein are very much dependent on the three-dimensional or tertiary structure.
Secondary Structure
Stretches or strands of proteins or peptides have distinct, characteristic local structural conformations, or secondary structure, dependent on hydrogen bonding. The two main types of secondary structure are the α-helix and the ß-sheet.
The α-helix is a right-handed coiled strand. The side-chain substituents of the amino acid groups in an α-helix extend to the outside. Hydrogen bonds form between the oxygen of each C=O bond in the strand and the hydrogen of each N-H group four amino acids below it in the helix. The hydrogen bonds make this structure especially stable. The side-chain substituents of the amino acids fit in beside the N-H groups.
The hydrogen bonding in a ß-sheet is between strands (inter-strand) rather than within strands (intra-strand). The sheet conformation consists of pairs of strands lying side-by-side. The carbonyl oxygens in one strand bonds with the amino hydrogens of the adjacent strand. The two strands can be either parallel or anti-parallel depending on whether the strand directions (N-terminus to C-terminus) are the same or opposite. The anti-parallel ß-sheet is more stable due to the more well-aligned hydrogen bonds.
Tertiary Structure
The overall three-dimensional shape of a protein molecule is the tertiary structure. The protein molecule will bend and twist in such a way as to achieve maximum stability or lowest energy state. Although the three-dimensional shape of a protein may seem irregular and random, it is fashioned by many stabilizing forces due to bonding interactions between the side-chain groups of the amino acids.
Under physiologic conditions, the hydrophobic side-chains of neutral, non-polar amino acids such as phenylalanine or isoleucine tend to be buried on the interior of the protein molecule, thereby shielding them from the aqueous medium. The alkyl groups of alanine, valine, leucine and isoleucine often form hydrophobic interactions between one another, while aromatic groups such as those of phenylalanine and tyrosine often stack together. Acidic or basic amino acid side-chains will generally be exposed on the surface of the protein as they are hydrophilic.
The formation of disulfide bridges by oxidation of the sulfhydryl groups on cysteine is an important aspect of the stabilization of protein tertiary structure, allowing different parts of the protein chain to be held together covalently. Additionally, hydrogen bonds may form between different side-chain groups. As with disulfide bridges, these hydrogen bonds can bring together two parts of a chain that are some distance away in terms of sequence. Salt bridges, ionic inter- actions between positively and negatively charged sites on amino acid side chains, also help to stabilize the tertiary structure of a protein.
Quaternary Structure
Many proteins are made up of multiple polypeptide chains, often referred to as protein subunits. These subunits may be the same, as in a homodimer, or different, as in a heterodimer. The quaternary structure refers to how these protein subunits interact with each other and arrange themselves to form a larger aggregate protein complex. The final shape of the protein complex is once again stabilized by various interactions, including hydrogen-bonding, disulfide-bridges and salt bridges. The four levels of protein structure are shown in Figure 2.
Protein Stability
Due to the nature of the weak interactions controlling the three-dimensional structure, proteins are very sensitive molecules. The term native state is used to describe the protein in its most stable natural conformation in situ. This native state can be disrupted by several external stress factors including temperature, pH, removal of water, presence of hydrophobic surfaces, presence of metal ions and high shear. The loss of secondary, tertiary or quaternary structure due to exposure to a stress factor is called denaturation. Denaturation results in unfolding of the protein into a random or misfolded shape.
A denatured protein can have quite a different activity profile than the protein in its native form, usually losing biological function. In addition to becoming denatured, proteins can also form aggregates under certain stress conditions. Aggregates are often produced during the manufacturing process and are typically undesirable, largely due to the possibility of them causing adverse immune responses when administered.
In addition to these physical forms of protein degradation, it is also important to be aware of the possible pathways of protein chemical degradation. These include oxidation, deamidation, peptide-bond hydrolysis, disulfide-bond reshuffling and cross-linking. The methods used in the processing and the formulation of proteins, including any lyophilization step, must be carefully examined to prevent degradation and to increase the stability of the protein biopharmaceutical both in storage and during drug delivery.
Protein Structure Analysis
The complexities of protein structure make the elucidation of a complete protein structure extremely difficult even with the most advanced analytical equipment. An amino acid analyzer can be used to determine which amino acids are present and the molar ratios of each. The sequence of the protein can then be analyzed by means of peptide mapping and the use of Edman degradation or mass spectroscopy. This process is routine for peptides and small proteins but becomes more complex for large multimeric proteins.
Peptide mapping generally entails treatment of the protein with different protease enzymes to chop up the sequence into smaller peptides at specific cleavage sites. Two commonly used enzymes are trypsin and chymotrypsin. Mass spectroscopy has become an invaluable tool for the analysis of enzyme digested proteins, by means of peptide fingerprinting methods and database searching. Edman degradation involves the cleavage, separation and identification of one amino acid at a time from a short peptide, starting from the N-terminus.
One method used to characterize the secondary structure of a protein is circular dichroism spectroscopy (CD). The different types of secondary structure, α-helix, ß-sheet and random coil, all have characteristic circular dichroism spectra in the far-UV region of the spectrum (190-250 nm). These spectra can be used to approximate the fraction of the entire protein made up of each type of structure.
A more complete, high-resolution analysis of the three-dimensional structure of a protein is carried out using X-ray crystallography or nuclear magnetic resonance (NMR) analysis. To determine the three-dimensional structure of a protein by X-ray diffraction, a large, well-ordered single crystal is required. X-ray diffraction allows measurement of the short distances between atoms and yields a three-dimensional electron density map, which can be used to build a model of the protein structure.
The use of NMR to determine the three-dimensional structure of a protein has some advantages over X-ray diffraction in that it can be carried out in solution and thus the protein is free of the constraints of the crystal lattice. The two-dimensional NMR techniques generally used are NOESY, which measures the distances between atoms through space, and COESY, which measures distances through bonds.
Protein Structure Stability Analysis
Many different techniques can be used to determine the stability of a protein. For the analysis of unfolding of a protein, spectroscopic methods such as fluorescence, UV, infrared and CD can be used. Thermodynamic methods such as differential scanning calorimetry (DSC) can be useful in determining the effect of temperature on protein stability. Comparative peptide-mapping (usually using LC/MS) is an extremely valuable tool in determining chemical changes in a protein, such as oxidation or deamidation. HPLC is also an invaluable means of analyzing the purity of a protein. Other analytical methods such as SDS-PAGE, iso-electric focusing and capillary electrophoresis can also be used to determine protein stability, and a suitable bioassay should be used to determine the potency of a protein biopharmaceutical. The state of aggregation can be determined by following “particle” size and arrayed instruments are now available to follow this over time under various conditions.
The variety of methods for determining protein stability again emphasizes the complexity of the nature of protein structure and the importance of maintaining that structure for a successful biopharmaceutical product.
References
- Protein Structure, Stability and Folding, Methods in Molecular Biology, 168, Edited by Kenneth P. Murphy
- Protein Stability and Folding, Theory and Practice, Methods in Molecular Biology, Vol. 40, Edited by Bret Shirley